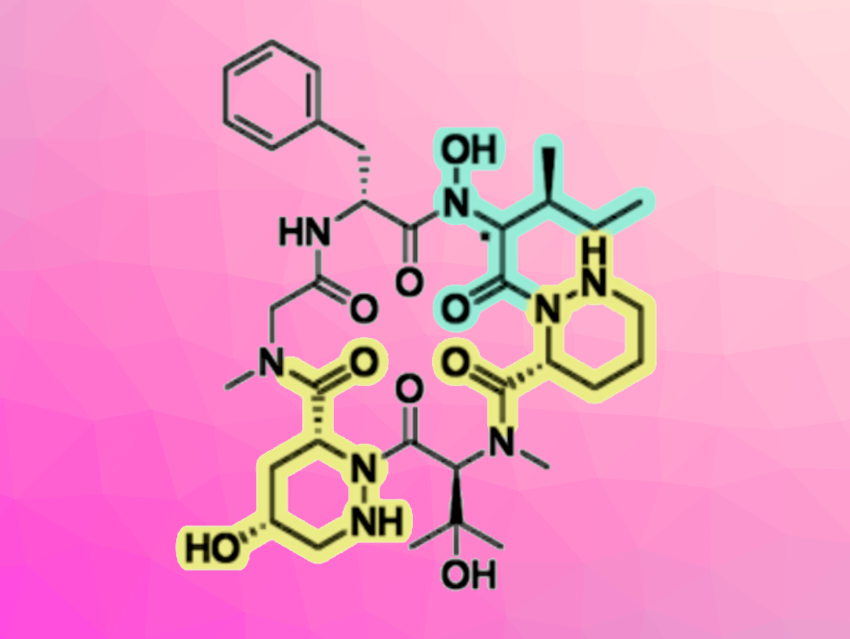
Pargamicin A is an 18-membered natural cyclopeptide containing unique non-proteinogenic piperazic acid derivatives. The compound has shown potent antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus faecalis/faecium (VRE). Thus, pargamicin A could be a good candidate for the development of new antibiotics for the treatment of infectious diseases, including those involving multi-drug resistant bacteria.
Masahito Yoshida, University of Tsukuba, Ibaraki, Japan, and colleagues have performed a straightforward total synthesis and structure determination of pargamicin A. The team used a convergent synthesis approach. They first prepared two key tripeptide segments using a linear peptide elongation process that involves the direct coupling of low-nucleophilic piperazic acids, using an in-situ-generated acid chloride derivative. The tripeptides were then coupled using triphosgene/collidine, giving a precursor for the cyclization step. Finally, a propylphosphonic anhydride (T3P)-mediated macrolactamization, followed by the removal of a benzyl protecting group, gave two putative structures of pargamicin A. When the team compared the properties of these diastereomers with the natural product, they found that the absolute configuration of the N-hydroxy-Ile residue of pargamicin A is 2S, 3S.
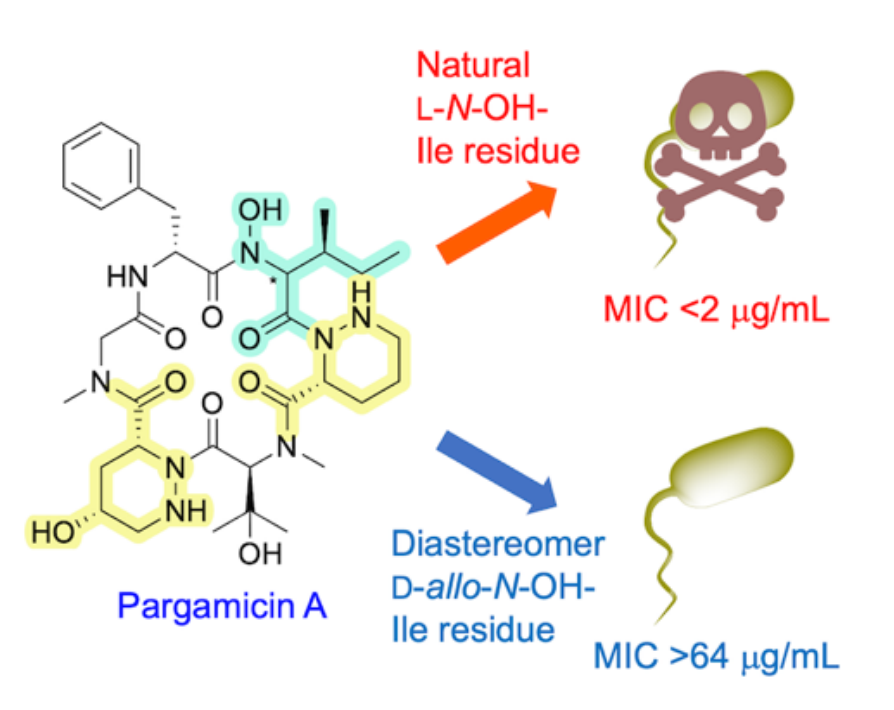
An evaluation of the antibacterial activities of the products showed that the synthetic pargamicin A exhibits similar potency to the natural product, but the diastereomer did not (pictured, MIC = minimum inhibitory concentration). These results suggest that the stereochemistry of the cyclopeptide scaffold can be crucial to induce desired antibacterial activity.
- Concise Total Synthesis and Biological Evaluation of Pargamicin A and its Diastereomer, Piperazic Acid‐containing Cyclopeptides,
Masahito Yoshida, Tetsuya Inaba, Yuko Shibuya, Masayuki Igarashi, Hideo Kigoshi,
ChemPlusChem 2023.
https://doi.org/10.1002/cplu.202300339
![]()



