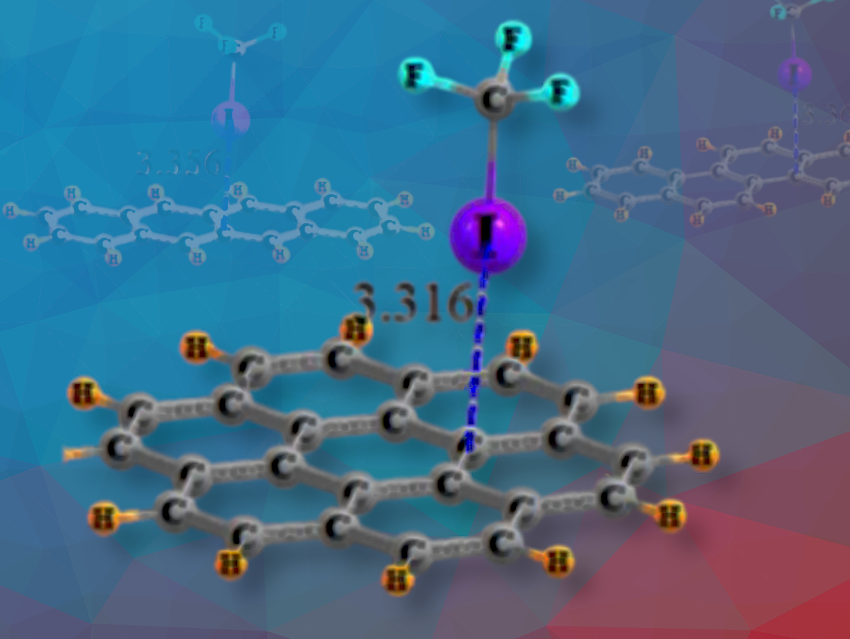
Steve Scheiner, a Professor at Utah State University, Logan, USA, and a colleague have used DFT calculations to study the tendency of π-electron systems in polycyclic aromatic compounds to form halogen bonds. In their study, they place CF3I, a prototype Lewis acid, above the aromatic planes of benzene, naphthalene, anthracene, phenanthrene, naphthacene, chrysene, triphenyl, pyrene, and coronene which function as Lewis bases.
A fairly strong CI··π halogen bond is formed between CF3I and any of a set of polycyclic aromatic systems, varying from the single ring of benzene to the seven rings of coronene. The iodine atom in the CF3I molecule positions itself a short distance above the surface of the aromatic compounds (3.3-3.4 Å) and shows a moderate interaction strength (4 kcal/mol). It is somewhat weaker with benzene than with the larger aromatic compounds.
The energy varies slightly as CF3I moves across the aromatic surface, favoring areas towards the sides of the rings with higher π-electron density. Although other forces like electrostatics also play a role, London dispersion is the most significant contributor to the binding energy between the halogen and the aromatic system. Therefore, the specific details of the molecular electrostatic potential (MEP) are not crucial for determining where the molecule attaches.
Why are these studies of interest to you?
The interest in the fundamental properties and numerous potential applications of halogen bonds continues to grow. Since hydrogen bonds are known to involve π-electron systems as donors, the obvious question is whether the closely parallel halogen bond can do so as well, and if so, how strong might such a halogen bond be?
What is new and cool about it?
Since both halogen atoms and aromatic systems of various sizes are prevalent in chemical and pharmaceutical systems, the ability of these two sorts of chemical moieties to bind to one another presents some outstanding opportunities for drug development. The results amplify the potential for these sorts of interactions to play a role in numerous chemical and biological phenomena.
What are your key findings?
The calculations revealed that halogen bonds can form and that their strength is comparable to other sorts of halogen bonds as well as hydrogen bonds. The strength of the bond is only slightly affected by the size of the polycyclic aromatic system.
The halogen donor can slide all the way across the face of the aromatic with little change in the interaction energy, providing ample opportunity for the halogen donor to interact even in crowded spaces.
What is the longer-term vision for your research?
The development of new pharmaceuticals can be aided by knowledge as to how a halogen-bearing group might interact with the aromatic systems, that are either part of a protein system, or by an internal halogen bond that controls the conformation of the pharmaceutical agent.
What part of your work was the most challenging?
There was a challenge first in choosing an appropriate quantum chemical method that can accurately model this interaction. The large size of some of the aromatics made it difficult to apply some of the most intrinsically accurate methods available.
How did you solve this problem?
High-level ab initio calculations such as CCSD(T) are not feasible for systems of this size. DFT offers an optimal compromise between accuracy and computational efficiency. The particular functional chosen has been shown to be reliable in several benchmark studies in the literature as well as in our own studies.
Thank you for these insights.
The article they talked about
- Halogen Bonding to the π-Systems of Polycyclic Aromatics,
Steve Scheiner, Akhtam Amonov,
ChemPhysChem 2024.
https://doi.org/10.1002/cphc.202400482

Steve Scheiner is a Professor at Utah State University, Logan, USA. (Photo: Utah State University)
![]()


