Efficacy Without Side Effects
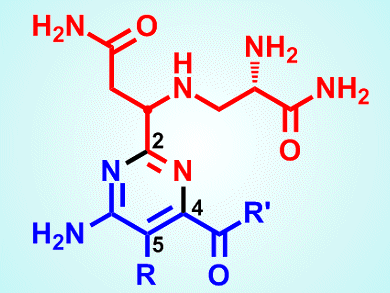
The total syntheses of two analogues of the compound bleomycin A2, (–)-pyrimidoblamic acid (see picture; R = CH3, R’ = OH) and P-3A (see picture; R = H, R’ = NH-L-His-L-Ala), by researchers at The Scripps Research Institute, La Jolla, CA, USA, could pave the way to finding a replacement for a common anticancer drug used for often devastating forms of the disease but avoiding its side effects.
Bleomycin A2, a glycopeptide antibiotic, anticancer agent produced by the bacterium Streptomyces verticillus and discovered in 1966, is used in the anticancer formulation Blenoxane against a range of conditions. These include Hodgkin’s lymphoma, melanoma, head and neck carcinomas, and testicular cancer. Its mode of action is derived from its ability to cleave DNA selectively, with double-strand breaks being the most important. Unfortunately, the drug’s efficacy is offset by the fact that it can give rise to dose-dependent pneumonitis and lung fibrosis, as well as having skin toxicity. These side effects are thought to be caused by the bithiazole group at the molecule’s C-terminal appendage, according to Adam Duerfeldt and Dale Boger.
Duerfeldt and Boger suggest that bleomycin-type compounds that lack this bithiazole group might be developed that target the DNA in cancer cells but without having the so-called “off-target” toxicity of the parent compound. The pair points out that each part of the parent structure has a role to play in its ability to cleave DNA, first through binding and thence to a cleavage cascade. Numerous systematic studies have pinned down the particular role of each subunit. The pyrimidoblamic acid moiety, for instance, is involved in metal chelation, oxygen activation, and thence triplex-like hydrogen bonding to guanine that trigger cleavage. Research has shown that removing the pyrimidine C5-methyl group does not affect activity detrimentally and they have therefore developed synthetic schemes to produce analogues that lack this feature.
Late-Stage Divergent Synthesis of Analogues
Previously, the team had developed a selective approach to cycloaddition for reacting substituted electron-deficient 1,2,3-triazines with various dienophiles and in particular a first demonstration of unknown [4 + 2] cycloaddition reactions with amidine or imidate heterodienophiles to give substituted pyrimidines. It was on these reactions that the team has developed second generation asymmetric total syntheses for (–)-pyrimidoblamic acid and P-3A. They explain that they were able to preload all the requisite stereochemistry before the [4 + 2] cycloaddition and introduction of the pyrimidine is to be carried out. This avoided some of the limitations of earlier routes, allowing them to create divergent analogues at a late stage without losing the reactivity needed for these final steps. The reactions occur at or around room temperature, which is always a convenience for a laboratory synthesis.
The team explains that, “In addition to permitting full control of the natural product stereochemistry, the approach provides the opportunity for the late-stage divergent synthesis of modified analogues bearing deep-seated changes in either the pyrimidine core (C4 and C5) or the highly functionalized C2-side chain.” They are currently working on developing the method further to create additional analogues for further investigation as putative anticancer drugs. They also point out that related heterocyclic groups are present in many other natural products and so this same approach might be adapted to the synthesis of a range of compounds.
- Total Syntheses of (−)-Pyrimidoblamic Acid and P-3A,
Adam S. Duerfeldt, Dale L. Boger,
J. Am. Chem. Soc. 2014.
DOI: 10.1021/ja412298c



