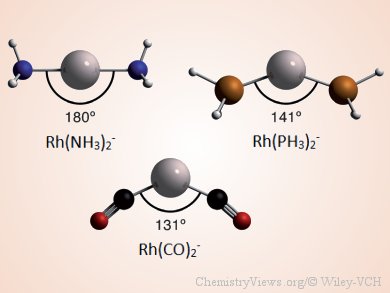
Lando P. Wolters and F. Matthias Bickelhaupt, VU University, The Netherlands, show that not all d10-ML2 complexes are linear. They investigated the molecular geometries and electronic structure of a series of d10-ML2 complexes (M = Co−, Rh−, Ir−, Ni, Pd, Pt, Cu+, Ag+, Au+; L = NH3, PH3, CO) by using relativistic density functional theory (DFT).
By analyzing the bonding mechanism in ML2 as a function of the ligand-metal-ligand (L-M-L) angle with quantitative Kohn–Sham molecular orbital (MO) theory in combination with an energy decomposition analysis (EDA) scheme, they show that the L-M-L angle can vary. Necessary for the occurrence of bent d10-ML2 complexes is sufficiently strong π backdonation. Smaller angles increase the overlap of the ligand’s acceptor orbital with a higher-energy donor orbital on the metal–ligand fragment and, therefore, favor π backdonation, resulting in additional stabilization. The balance between the additional stabilization and the increased steric repulsion, occurring as the complexes are bent, determines the resulting angle of the complex.
The analyses explain the phenomenon and provide a tool for rationally tuning the bite angle.
 Nonlinear d10-ML2 Transition-Metal Complexes
Nonlinear d10-ML2 Transition-Metal Complexes
Lando P. Wolters, Prof. Dr. F. Matthias Bickelhaupt
ChemistryOpen 2013, 3, 106–114,
DOI: 10.1002/open.201300009


