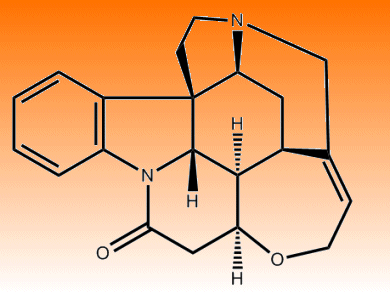
Small Molecule, Complex Structure
Strychnine in pure form is colorless, incredibly bitter and rather toxic. Just 80 milligrams is enough to cause the agonizing death of an 80 kg adult. The alkaloid was first isolated in 1818 and had a bipolar relationship with medicine – on the one hand it was a useful laxative, but a minor overdose caused convulsions and a nudge of the pharmacist’s scales would be the patient’s demise. In 1946, renowned chemist Sir Robert Robinson together with Herman Leuchs determined its structure and proclaimed that, “For its molecular size, it is the most complex substance known.”
Small molecule, complex structure? That could be the most enticing phrase an organic chemist might hear, in the laboratory at least. As such, countless synthetic schemes have been devised to convert other simpler small molecules into the elaborate ring system of strychnine. The nightmarish effects of strychnine are, however, nothing compared to its systematic name, (1R,11S,18S,20R,21R,22S)-12-Oxa-8,17-diazaheptacyclo[15.5.2.01,18.02,7.08,22.011,21.015,20]tetracosa-2,4,6,14-tetraen-9-one, which hints at just how difficult finding a succinct pathway to this compound has been.
Total Synthesis – From 21 to 6 Steps
The classic total synthesis of strychnine was published in 1954 by Nobel chemist Robert Burns Woodward [1]. During that golden era of natural product synthesis, Woodward also synthesized quinine, cholesterol, cortisone, lysergic acid, reserpine, chlorophyll, cephalosporin and colchicine, and pioneered the modern field of synthetic organic chemistry. Larry Overman abbreviated the route to strychnine, cutting it down to 18 reaction steps [2]. Woodward’s scheme required 21. Some of the greatest names in twentieth century chemistry and beyond have looked to strychnine for synthetic inspiration and devised novel approaches and useful side-reactions in their efforts to reach the final product in as few steps as possible.
Shibasaki achieved it in 15 steps [3], and others including Vollhardt, Mori, Fukuyama, Kuehne, Bosch, Rawal (14 steps, the previous record) and others have trimmed the reactions, boosted the yields, avoided the side products and generally moved towards a faster, shorter and more efficient synthesis. Now, an astoundingly concise six-step reaction scheme has been published by Christopher Vanderwal and his colleagues at the University of California, Irvine, USA [4]. The team starts with commercially available materials and applies some old and some new chemistry to metamorphose those materials into strychnine.
“The synthesis proceeds in six steps via the longest linear sequence from any commercially available starting material,” explains Vanderwal, “so the total number of steps is more like ten or so. But the longest linear sequence is what most people are comparing, so the number six is quite relevant.”
Zincke Aldehyde Approach
Vanderwal and colleagues explain how two previous approaches to strychnine have successfully exploited the Diels-Alder cycloaddition reaction. However, the researchers felt that their chemical prowess in using a tryptamine-derived Zincke aldehyde to create a chemical cousin of strychnine, the natural product norfluorocurarine, might lend itself to a shorter synthesis to the archetypal toxin. “Our strategy used in our previous synthesis of the simpler Strychnos alkaloid norfluorocurarine suggested a way to make strychnine in a very short way,” Vanderwal told us, “The precursor to strychnine that we aimed for in this paper is the Wieland–Gumlich aldehyde.”
They point out that the Zincke aldehyde approach positions appropriate functional groups and leaves them in the correct oxidation states for the final steps. They point out, with apparent pride, how a mere four chemical steps creates four new carbon-carbon bonds and one carbon-oxygen bond to make a five-carbon donor-acceptor diene.
The team concedes that the final yield was not quite as high as they had hoped, however, they point out that the strategy in general demonstrates such effect in bond creation and setting up the intermediate in very few steps that the same chemistry will be very useful in targeting related structures and their derivatives anyone of which might have physiological activity and possible medicinal benefits beyond those of strychnine. There are, of course, many more complex synthetic targets on which whole chemistry careers have been built, but none seems to have quite the fatal attraction of strychnine.
- The total synthesis of strychnine
R. B. Woodward, Michael P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker, K. Schenker,
J. Am. Chem. Soc. 1954, 76, 4749.
DOI: 10.1021/ja01647a088 - Synthesis applications of cationic aza-Cope rearrangements. 26. Enantioselective total synthesis of (-)-strychnine
S. D. Knight, L. E. Overman, G. Pairaudeau,
J. Am. Chem. Soc. 1993, 115(20), 9293–9294.
DOI: 10.1021/ja00073a057 - Enantioselective Total Synthesis of (−)-Strychnine Using the Catalytic Asymmetric Michael Reaction and Tandem Cyclization
T. Ohshima, Y. Xu, R. Takita, S. Shimizu, D. Zhong, M. Shibasaki,
J. Am. Chem. Soc. 2002, 124(49), 14546–14547.
DOI: 10.1021/ja028457r - A synthesis of strychnine by a longest linear sequence of six steps
D. B. C. Martin, C. D. Vanderwal,
Chem. Sci. 2011.
DOI: 10.1039/c1sc00009h


![Synthesis of [c2]Daisy Chains via Mechanochemistry](https://www.chemistryviews.org/wp-content/uploads/2025/04/202504_RotaxanesWithSolidStateMechanochemistry-125x94.png)