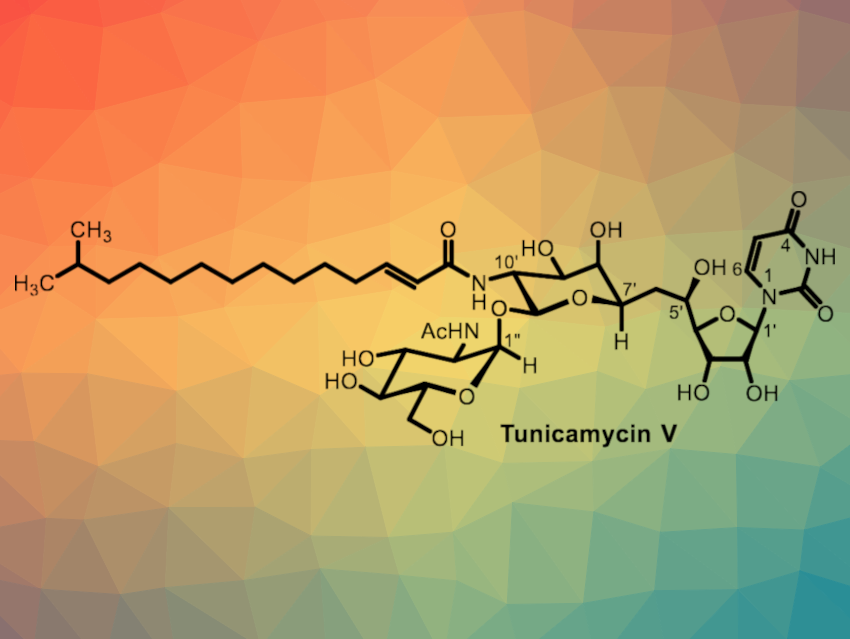
The tunicamycins are natural products that show cytotoxic activity against both cancer and normal cells in a non-selective manner. They have been used in research, but are difficult to advance into clinically useful drugs because of their low selectivity against the enzyme they inhibit, DPAGT1. This enzyme is important for N-glycan biosynthesis. A convergent and concise total synthesis of tunicamycin analogues would be useful and could allow researchers to study structure–activity relationships and minimize unwanted toxic effects.
Michio Kurosu, University of Tennessee Health Science Center, Memphis, USA, and colleagues have developed an efficient total chemical synthesis of tunicamycin V (pictured) via a Büchner–Curtius–Schlotterbeck-type reaction. The Büchner–Curtius–Schlotterbeck reaction is a reaction of aldehydes or ketones with aliphatic diazoalkanes. The synthesis features a selective 11′-β-1”-α-trehalose-type linkage, a selective mono N-nitrosylation of a diacetamide, and a stereoselective reduction of the C5′-carbonyl group. It requires only 15 chemical steps from D-galactal.
This short synthetic route was adapted and used for the synthesis of tunicamycin analogues. Using this approach, the team discovered a new cytostatic molecule with high DPAGT1 selectivity. They found that this cytostatic tunicamycin analogue was selective against certain cancer cell lines, including a triple-negative breast cancer.
- Concise Synthesis of Tunicamycin V and Discovery of a Cytostatic DPAGT1 Inhibitor,
Katsuhiko Mitachi, David Mingle, Wendy Effah, Antonio Sánchez-Ruiz, Kirk E. Hevener, Ramesh Narayanan, William M. Clemons, Francisco Sarabia, Michio Kurosu,
Angew. Chem. Int. Ed. 2022.
https://doi.org/10.1002/anie.202203225
![]()