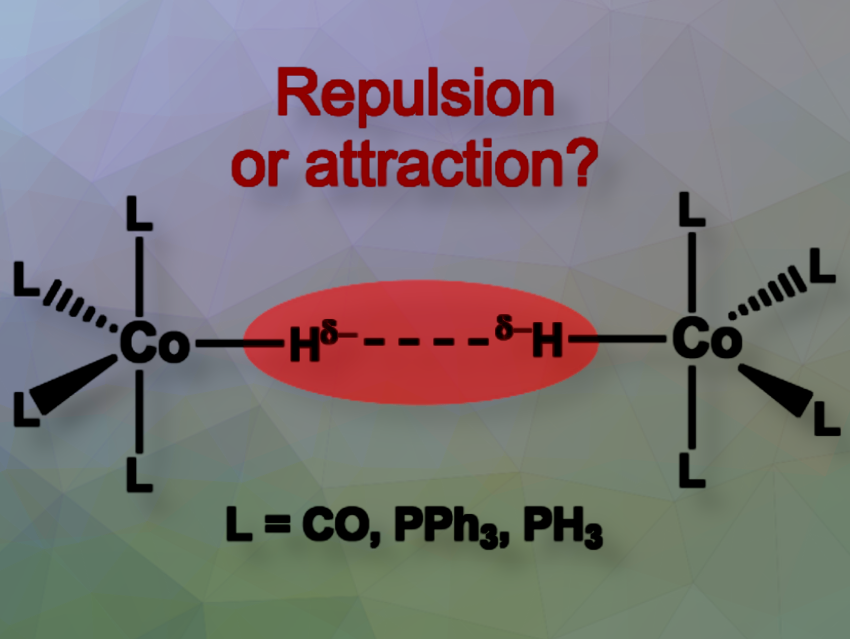
Mariusz Mitoraj, Jagiellonian University, Krakow, Poland, and colleagues have investigated hydride-hydride interactions (Hδ−···δ−H) in hydrogen storage materials. The high content of these contacts is relevant to hydrogen formation. Exploring such interactions in transition metal complexes like HCoL4 can help improve hydrogen storage by optimizing ligands, configurations, and interaction strengths.
What did you do?
We applied advanced quantum chemistry methods to explore the nature of intermolecular hydride-hydride (Hδ−···δ−H) interactions in hydrogen storage materials. Our study focused on well-known transition metal complexes, including cobalt tetracarbonyl hydride and its phosphine derivatives; HCoL4 where L = CO, PPh3, PH3.
By applying modern bonding descriptors, we analyzed whether these interactions can be attractive, contrary to intuitive wisdom, and identified their physical origin.
Why are you doing this?
Homopolar dihydrogen interactions remain a topic of debate, with no consensus on whether these contacts are attractive or repulsive. Our research aims to provide fundamental insights into these interactions, enhancing our understanding of the structure as well as the mechanism of dehydrogenations.
What is new and cool about it?
Our study challenges classical chemical intuition by revealing that homopolar hydride-hydride interactions can be attractive, and even stronger than classical hydrogen bonds. The strength varies based on ligand type and molecular conformation, with phosphine ligands increasing stability. Overall, Hδ−···δ−H bonding energy changes in the order: CO << PPh3 ~ PH3.
Moreover, these interactions show a chameleon-like behavior, varying from attractive to repulsive depending on the metal coordination. It is attractive in CO4CoH···HCoCO4, whereas repulsion is shown in (CO)3(PPh3)CoH···HCo(CO)3(PPh3). This unexpected adaptability adds a fascinating complexity to our understanding of chemical bonding. Furthermore, such attractive contacts might initiate dihydrogen release from metal hydrides.
What are your key findings?
Our research sheds new light on the “old topic” of seemingly repulsive hydride-hydride interactions. We discovered that depending on the monomers modifications, Hδ−···δ−H bonds can be attractive and dominated by charge delocalization or London dispersion forces, with electrostatic interactions also playing a crucial role.
Additionally, these dimers gain further stability from interactions with adjacent carbonyl ligands and other weak contacts, such as π···π stacking, C−H···π, and C−H···H−Co.
What is the longer-term vision for your research?
Our future research will focus on exploring the dynamics of these systems at various temperatures to understand their behavior over time, particularly in relation to the dehydrogenation mechanisms. Since cobalt tetracarbonyl hydride is known for its volatility and instability due to H₂ elimination, we conclude that attractive hydride-hydride interactions might play a crucial role in driving the dehydrogenation processes.
These insights have important implications for hydrogen storage technologies, as they could lead to the development of more efficient materials for hydrogen storage, contributing to the advancement of clean energy solutions.
What part of your work was the most challenging?
The most challenging aspect was to find the most reliable and accurate methodology by studying various exchange-correlation functionals of the Jacob’s ladder, followed by applications of wave-function-based approaches. Jacob’s ladder refers to a hierarchy of increasingly accurate exchange-correlation functionals in density functional theory (DFT). We used this framework to find the most reliable computational method for analyzing hydride-hydride interactions followed by applying more precise wave-function-based approaches together with various bonding descriptors: the Extended Transition State-Natural Orbital for Chemical Valence-method (ETS-NOCV), Interacting Quantum Atoms (IQA), and reduced density gradient (NCI).
Additionally, selecting the right systems was necessary, which has been done by extensive analyses of hydride dissociation constants and the electronic structure of metal complexes.
Finally, reactivity initiated by hydride-hydride interactions now constitutes a crucial subject in the context of homogeneous and heterogeneous dihydrogen production.
Thank you for these insights.
The paper they talked about:
- Theoretical description of hydride-hydride interactions in selected hydrogen storage materials,
Mariusz Pawel Mitoraj, Aleksandra L. Ptaszek, Filip Sagan, Radosław Filas, Piotr Kubisiak,
ChemPhysChem 2024.
https://doi.org/10.1002/cphc.202400668
Mariusz Mitoraj, is Professor at the Department of Theoretical Chemistry in the Faculty of Chemistry at Jagiellonian University in Krakow, Poland




