Macrocyclic peptides are useful components of several pharmaceuticals. However, their creation is challenging because intermolecular reactions can generate unwanted dimers and oligomers. Conventional macrocyclization reactions are, thus, carried out at high dilution and with long reaction times to promote ring-closing reactions instead of these side reactions.
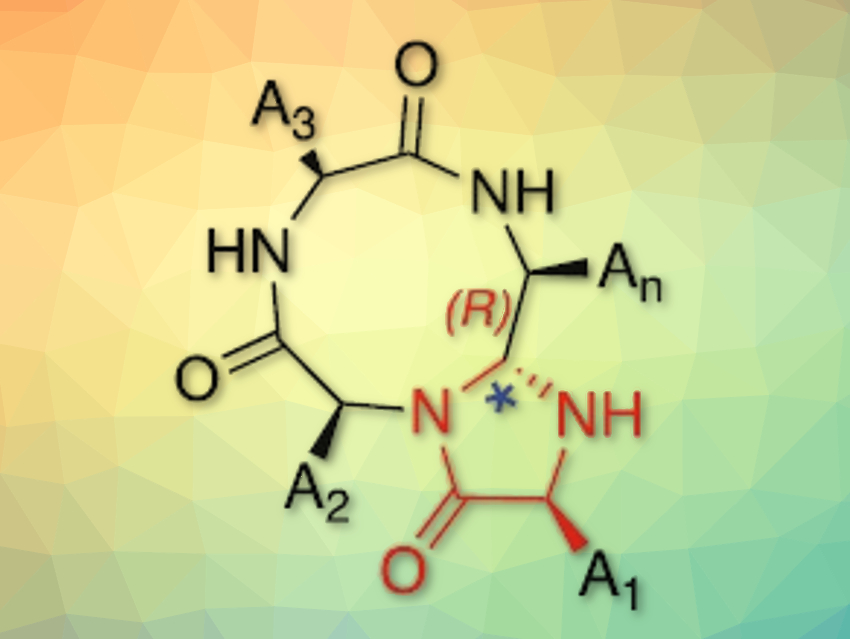
Monika Raj, Auburn University, AL, USA, and colleagues have developed a method for the synthesis of macrocyclic peptides. The so-called “CyClick” reaction is selectively intramolecular and creates cyclic peptides in high yields without side products. The cyclization of linear peptides is performed in a 1:1 mixture of water and N,N-dimethylformamide (DMF) in the presence of 4-dimethylaminopyridine (DMAP) at room temperature. The reaction first gives a cyclic imine intermediate (pictured in the center below), which undergoes another ring-closing to form a stable 4-imidazolidinone-fused cyclic peptide A new chiral center is generated in this process with high stereoselectivity.
.jpg)
The CyClick reaction can be carried out in one pot and under mild conditions. A variety of cyclic peptides with different ring sizes (12- to 24-membered) and amino acid compositions are accessible. High conversions are reached even at 25 times the concentration of current methods. The researchers believe that this process could streamline the synthesis of macrocyclic peptides for pharmaceutical applications.
- CyClick Chemistry for the Synthesis of Cyclic Peptides,
Victor Adebomi, Ryan D. Cohen, Rachel Wills, Holland Andrew Hays Chavers, Gary E. Martin, Monika Raj,
Angew. Chem. Int. Ed. 2019.
https://doi.org/10.1002/anie.201911900
![]()