Marine Antitumor Agents
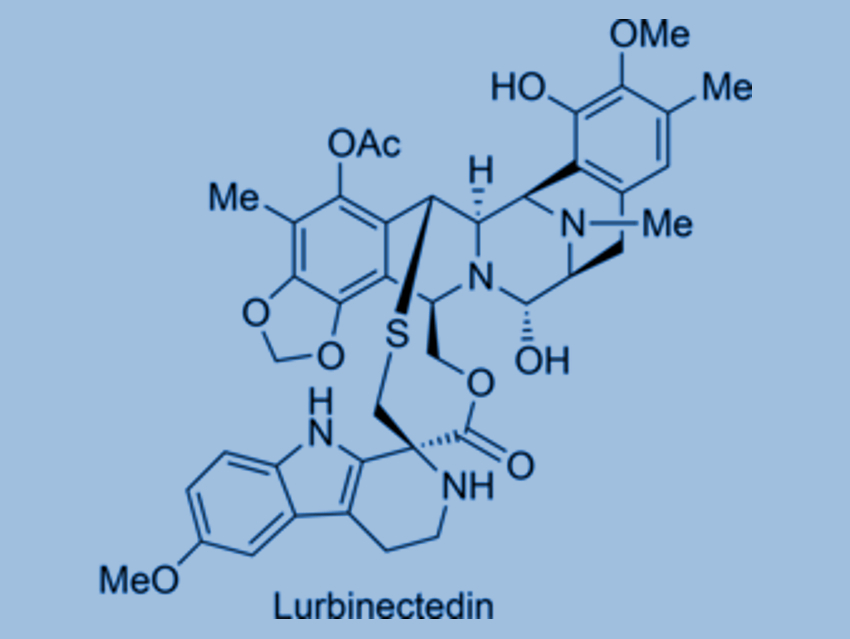
With its vast numbers of different lifeforms, the sea is a largely unexplored source of natural products that could be starting points for new pharmaceuticals, such as the antitumor drugs trabectedin and lurbinectedin (pictured). Because only tiny amounts can be obtained from sea organisms, synthetic production is necessary. Dawei Ma, University of the Chinese Academy of Sciences, Shanghai, China, and colleagues have developed a new, efficient synthetic route for these two drugs. A key step is the light-controlled activation of a carbon–hydrogen bond.
Trabectedin (also called ecteinascedin) comes from the sea squirt species Ecteinascidia turbinata and is the first marine natural product to be used clinically as a drug—for the treatment of advanced soft tissue sarcoma. Lurbinectedin has a slightly modified structure and is currently in phase III clinical studies for the treatment of certain lung and breast cancers.
Total Synthesis of Trabectedin and Lurbinectedin
One ton of sea squirts is needed to acquire about one gram of trabectedin. A viable and efficient synthetic route for making this and related drugs in sufficient quantities is therefore needed urgently. However, trabectedin has thus far proven to be one of the most challenging target molecules in natural products synthesis. Various synthetic routes have been proposed but none is really viable. Current methods are very complex, require expensive and uncommon reagents, and deliver unsatisfactory yields.
The researchers have developed a more efficient and viable de novo synthetic route for trabectedin and lurbinectedin. The synthesis starts with the amino acid S-tyrosine and consists of 26 individual steps. First, several steps are used to produce an intermediate which acts as the starting material for the separate production of the two halves of the target molecule—trabectedin or lurbinectedin. These are then bound together in a later reaction step.
Light-Controlled Activation of a C–H Bond
The key step of the synthesis is the light-controlled activation of a normally unreactive carbon–hydrogen bond (remote C–H activation). A radical rearrangement mechanism leads to a ring closure in which a quinone group is converted into a 1,3-benzodioxole unit, which is a structural component found in many natural products. The reaction was particularly efficient under irradiation with blue light in tetrahydrofuran (THF) as a solvent.
The scientists hope that their synthetic route offers a practical and economical method for the production of trabectedin and lurbinectedin, finally providing adequate supplies of these complex marine antitumor drugs.
- A Scalable Total Synthesis of the Antitumor Agents Et-743 and Lurbinectedin,
Weiming He, Zhigao Zhang, Dawei Ma,
Angew. Chem. Int. Ed. 2019.
https://doi.org/10.1002/anie.201900035