Theoretical and computational chemistry tools are routinely used to effectively design and discover new drugs. The binding energy of the drug (or ligand) to the protein of interest is one key parameter in these analyses. This calculation requires numerous parameters to be optimized, including desolvation penalties and dynamic conformational corrections. An alternative approach is to measure the changes in binding energy of a known ligand–protein complex when the substituents of the ligand are changed.
Daniel L. Cheney, Bristol-Myers Squibb Company, Pennington, NJ, USA, C. David Sherrill, Georgia Institute of Technology, Atlanta, GA, USA, and colleagues have used their computational method called functional-group symmetry-adapted perturbation theory (F-SAPT) to calculate the strength of the noncovalent contacts between functional groups of a ligand–protein complex. This includes contributions of electrostatics, polarization, London dispersion, and exchange-repulsion interactions.
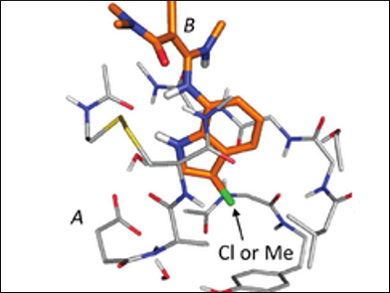
Using this method, the team looked at the change in the interaction energy between a chloro- and a methyl-substituted ligand of factor Xa (fXa), a key enzyme in blood coagulation (pictured). The researchers showed that the difference in the interaction energy of the two ligands with fXa was 2.46 kcal/mol, which is mainly due to favorable electrostatic interactions between the chloro-substituted ligand and the peptide bonds (backbone) in the binding pocket.
The study indicates that interactions based on a visual identification of various interaction motifs like hydrogen bonds may not always be sufficient to understand differences between chemically similar drugs. Large-scale models of the ligand and protein binding pocket, using F-SAPT, among others, may be necessary to understand the change in the interaction energy.
- The Surprising Importance of Peptide Bond Contacts in Drug-Protein Interactions,
Robert M. Parrish, Doree F. Sitkoff, Daniel L. Cheney, C. David Sherrill,
Chem. Eur. J. 2017, 23, 7887–7890.
DOI: 10.1002/chem.201701031